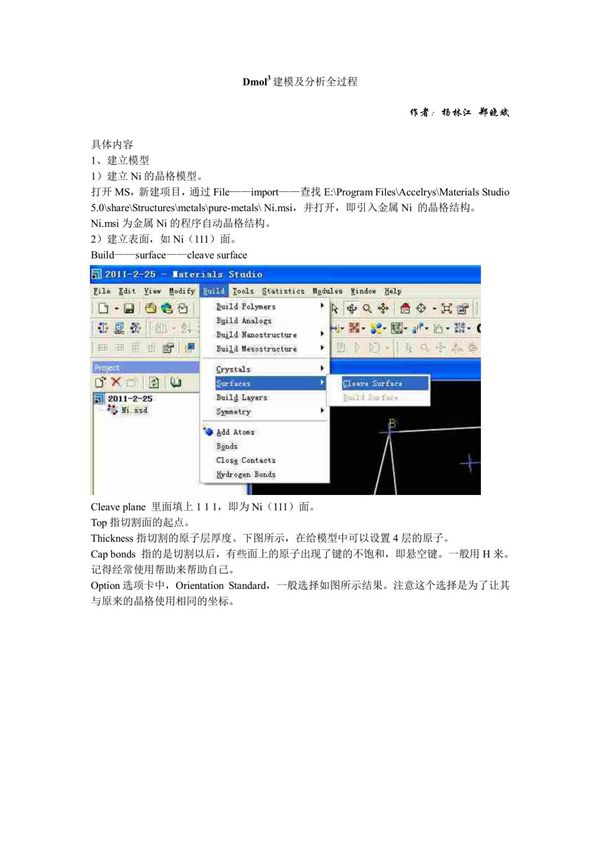
Dmol3是一款基于密度泛函理论(DFT)的量子力学计算软件,广泛应用于材料科学、化学和物理等领域。它能够高效地进行分子和周期性体系的电子结构计算、几何优化以及多种性质分析。Dmol3建模及分析优化的全过程通常包括以下几个关键步骤:1.**模型构建**:根据研究体系(分子、表面、晶体等)创建初始结构,可通过手动搭建或导入已有结构文件(如.cif、.xyz等格式)。2.**参数设置**:选择适当的泛函(如GGA-PBE)、基组(如DNP)、计算精度(如Medium或Fine)以及自洽场(SCF)收敛标准。3.**几何优化**:对初始结构进行能量最小化计算,调整原子位置以获得稳定构型,确保力收敛和能量收敛标准得到满足。4.**性质计算**:优化完成后,可进一步计算电子结构(如能带、态密度)、光学性质、振动频率或反应能垒等。5.**结果分析**:通过可视化工具(如MaterialsStudio)检查优化结构、电子分布、能量变化等,验证计算结果的合理性。Dmol3以其高效的计算性能和友好的用户界面,成为研究材料电子性质及反应机理的重要工具。